The SARS-CoV-2 spike protein disrupts the cooperative function of human cardiac pericytes – endothelial cells through CD147 receptor-mediated signalling: a potential non-infective mechanism of COVID-19 microvascular disease
Abstract
Background Severe coronavirus disease 2019 (COVID-19) manifests as a life-threatening microvascular syndrome. The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) uses primarily the capsid spike (S) protein to engage with its receptors and infect host cells. To date, it is still not known if the S protein alone, without the other viral elements, is able to trigger vascular cell signalling and provoke cell dysfunction.
Methods We investigated the effects of the recombinant, stabilised S protein on primary human cardiac pericytes (PCs) signalling and function. Endpoints included cell viability, proliferation, migration, cooperation with endothelial cells (ECs) in angiogenesis assays, and release of pro-inflammatory cytokines. Adopting a blocking strategy against the S protein receptors ACE2 and CD147, we explored which receptor mediates the S protein signalling in PCs.
Findings We show, for the first time, that the recombinant S protein alone elicits functional alterations in cardiac PCs. This was documented as: (1) increased migration, (2) reduced ability to support EC network formation on Matrigel, (3) secretion of pro-inflammatory molecules typically involved in the cytokine storm, and (4) production of pro-apoptotic factors responsible for EC death. Furthermore, the S protein stimulates the phosphorylation/activation of the extracellular signal-regulated kinase 1/2 (ERK1/2) through the CD147 receptor, but not ACE2, in cardiac PCs. Accordingly, the neutralization of CD147, using a blocking antibody, prevented the activation of ERK1/2 and partially rescued the PC function in the presence of the S protein.
Interpretation Our findings suggest the new, intriguing hypothesis that the S protein may elicit vascular cell dysfunction, potentially amplifying, or perpetuating, the damage caused by the whole coronavirus. This mechanism may have clinical and therapeutic implication.
Funding Elizabeth Blackwell Institute (EBI) Rapid Response COVID-19 award.
Evidence before this study The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) uses primarily the capsid spike (S) protein to engage with its receptors and infect host cells. Co-receptors and host cell proteases may also be involved. Angiotensin-converting enzyme 2 (ACE2) is the well-recognized entry receptor used by the virus in respiratory epithelial cells; it is also abundantly expressed in the human heart. Alongside ACE2, CD147 has recently emerged as a novel receptor for SARS-CoV-2. Yet, it is not clear if SARS-CoV-2 triggers adverse responses in cardiac vascular mural cells. Likewise, no investigation was devoted to verifying if the recombinant S protein alone can mimic the whole virus signalling.
Added value of this study This study provides the first evidence that the recombinant S protein alone, without the other viral elements, is capable of eliciting cellular signalling in human cardiac pericytes, thereby inducing cell dysfunction. In addition, this study proposes CD147 as the leading receptor mediating S protein signalling in cardiac pericytes.
Implications of all the available evidence These reports imply that fragments of the S protein might be able to elicit vascular cell dysfunction. Blocking the CD147 receptor may help protect the vasculature not only from infection, but also from the collateral damage caused by the S protein.
Introduction
Severe coronavirus disease 2019 (COVID-19) is considered a microvascular disorder in which the infectious agent harms endothelial cells (ECs), causing inflammation and thrombosis.1 Thromboembolic events resulting in stroke or myocardial infarction occur in up to 4% of patients with COVID-19 hospitalized in intensive care units. Moreover, people with pre-existing cardiovascular disease are more likely to die of COVID-19.2
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) uses the homotrimeric spike (S) glycoprotein imbedded in the virus to bind to cognate receptors on human cells. Such binding triggers a cascade of events that leads to fusion of the viral and cellular membranes to facilitate virus entry,3 and subsequent manipulation of the host Raf/MEK/ERK signalling pathway to regulate viral replication and gene transcription in host cells.4 The receptor-binding domain (RBD) contained in the S1 subunit of the viral S protein recognizes and binds to the host receptor angiotensin-converting enzyme 2 (ACE2), while the S2 subunit mediates viral – cell membrane fusion by forming a six-helical bundle via the two-heptad repeat domain.5,6 Interestingly, the recombinant S1 subunit (Val16 – Gln690), but not the S1 subunit (Arg319 – Phe541) that contains the ACE2 RBD, induces MEK phosphorylation in pulmonary vascular cells.7 This finding suggests that the viral protein can induce intracellular signalling through alternative receptors independently of ACE2. Accordingly, another report indicated the S2 subunit to be responsible for disturbing the barrier function of brain ECs.8
CD147, also known as Basigin or extracellular matrix metalloproteinase inducer (EMMPRIN), has recently emerged as a novel receptor for SARS-CoV-2.9 This transmembrane protein is expressed by ECs, signals through ERK1/2, is upregulated during inflammation and atherothrombosis, and may contribute to plaque instability by inducing metalloproteinase expression.10 Therefore, CD147 represents a potential mediator of the cardiovascular damage caused by SARS-CoV-2. It remains, however, unknown whether SARS-CoV-2 infects and harms vascular cells of the heart through a mechanism involving ACE2 (the recognition of elevated ACE2 mRNA transcript levels in coronary mural cells suggested this possibility), 11,12 or Basigin/CD147, or both.
Pericytes (PCs) exert pleiotropic functions, supporting the integrity of coronary artery ECs (CAECs),13,14 participating in cardiac repair,15 and modulating inflammatory responses.16 Dysfunctional PCs participate in adverse vascular phenomena; for instance, after a heart attack, persistently contracted PCs block the microvascular coronary circulation, thereby causing blood to clot.17 Therefore, it is crucial to understand the role of these perivascular cells in COVID-19.
The present study aimed to answer the following questions: (1) Is CD147 expressed in human cardiac PCs? (2) Is the S protein sufficient to trigger molecular, functional, and pro-inflammatory alterations in cardiac PCs? (3) Can these changes be inhibited by shielding PCs with a CD147 neutralizing antibody?
Methods
Where not otherwise stated, all chemicals were purchased from Sigma-Aldrich.
Ethics
This study complies with the ethical guidelines of the Declaration of Helsinki. Human myocardial samples (right ventricle or atrium) were discarded material from surgical repair of congenital heart defects (ethical approval number 15/LO/1064 from the North Somerset and South Bristol Research Ethics Committee). Adult patients and paediatric patients’ custodians gave informed written consent. Donors and samples characteristics are described in Table 1. All patients were recruited before the COVID-19 pandemic.
Derivation of primary cultures of cardiac PCs
Immunosorted CD31negative/CD34positive PCs were expanded in a dedicated medium (ECGM2, C-22111, PromoCell) and confirmed to express a panel of typical markers using immunocytochemistry and flow cytometry, as previously described.13,18
Culture of cardiac fibroblasts and ECs
Human cardiac fibroblasts were purchased from PromoCell and expanded in Fibroblast Growth Medium 2 (FGM2, C-23120, PromoCell) according to manufacturer guidelines. Human CAECs were purchased from PromoCell and expanded in full Endothelial Cell Growth MicroVascular medium 2 (ECGMV2, C-22120, PromoCell) according to manufacturer’s guidelines.
Immunocytochemistry analysis
Cells were rinsed with PBS and fixed with 4% w/v PFA in PBS for 15 min at 20°C. After washing with PBS, the cells were permeabilized with 0·1% v/v Triton-X100 in PBS for 5 min at 20°C. Cells were blocked with 10% v/v FBS and incubated with antibodies anti-ACE2 (R&D AF933, goat polyclonal, dilution 1:50) or anti-CD147 (BioLegend #306221, mouse monoclonal, dilution 1:100), for 16 hrs at 4°C. Secondary antibodies conjugated with Alexa 488 were purchased from ThermoFisher Scientific and used at a dilution of 1:200, for 1 h at 20°C, in the dark. Nuclei were counterstained using Hoechst (1:10,000 in PBS, 3 min at 20°C). Cells were mounted using Fluoromount for imaging.
Gene expression analysis by real-time qPCR
Extracted total RNA was reverse-transcribed into single-stranded cDNA using a High-Capacity RNA-to-cDNA Kit (ThermoFisher Scientific). The RT-PCR was performed using first-strand cDNA with TaqMan Fast Universal PCR Master Mix (ThermoFisher Scientific). TaqMan primer-probes were obtained from ThermoFisher Scientific (ACE2 Hs05627131_gH; BSG Hs00936295_m1; housekeeping gene UBC Hs00824723_m1). Quantitative PCR was performed on a QuantStudio™ 5 System (ThermoFisher). All reactions were performed in a 10 μL volume in triplicate, using 7.5 ng cDNA per reaction. The mRNA expression levels were normalized against UBC and determined using the 2−ΔΔCt method.19
Production and purification of recombinant SARS-CoV-2 S protein
SARS-CoV-2 S protein was expressed in insect cells and purified as described before20,21 Briefly, the S construct consisted of amino acids 1 to 1213 fused with a thrombin cleavage site followed by a T4-foldon trimerization domain and a hexahistidine affinity purification tag at C-terminus. The polybasic furin cleavage site was mutated (RRAR to A) to increase the stability of the protein for in vitro studies.20,21 S protein was expressed in Hi5 cells using the MultiBac system.22 Secreted S protein was harvested 3 days after infection by centrifuging the cell culture at 1,000g for 10 min followed by another centrifugation of supernatant at 5,000g for 30 min. S protein-containing medium was incubated with HisPur Ni-NTA Superflow Agarose (Thermo Fisher Scientific) for 1h at 4°C. Resin bound with S protein was separated from unbound proteins and medium using a gravity flow column, followed by 30 column volume wash with wash buffer (65 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 7.5). Finally, the protein was eluted with a step gradient of elution buffer (65 mM NaH2PO4, 300mM NaCl, 235mM imidazole, pH 7·5). Eluted fractions were analyzed by reducing SDS-PAGE. Fractions containing the S protein were pooled and concentrated using 50 KDa MWCO Amicon centrifugal filter units (EMD Millipore). During concentration, proteins were buffer-exchanged in phosphate-buffered saline (PBS) pH 7.5. Concentrated protein was aliquoted, flash frozen in liquid nitrogen, and stored at -80°C until use.
Western blotting on total cell lysates
Whole-cell protein lysates were prepared using RIPA buffer supplemented with 1:50 proteases inhibitors cocktail and 1:100 phosphatases inhibitors. Cell lysates were centrifuged at 10,000 g, at 4°C, 15 min. After the assessment of protein concentration (BCA Protein Assay Kit, ThermoFisher Scientific), the supernatants were kept at -80°C. Twenty μg of protein lysates were prepared in Laemmli loading buffer, incubated for 8 min at 98°C, resolved on 10% SDS-PAGE, and transferred onto 0·2 μm PVDF or nitrocellulose membranes (Bio-Rad). Membranes were blocked using 5% w/v non-fat dried milk (Bio-Rad) in Tris-buffered saline (TBS, BioRad) supplemented with 0.05% v/v Tween-20 for 2 h at 20°C. Primary antibodies (ACE2, dilution 1:100; CD147, 1:500, 6x-HIS-tag (Invitrogen MA1-21315), 1:1000) were incubated for 16 hrs at 4°C. β-Actin was used as a loading control (Sigma, A5441, 1:10000). Anti-mouse IgG HRP (1:5000, GE Healthcare) or anti-goat IgG HRP (R&D, HAF017, 1:5000) conjugated antibodies were employed as secondary antibodies. Membrane development was performed by an enhanced chemiluminescence-based detection method (ECL™ Prime Western Blotting Detection Reagent, GE Healthcare) and observed using a ChemiDoc-MP system (Bio-Rad). Western blot data were analyzed using the ImageJ software.
For detection of the S protein binding to PCs, 1 μg/mL (5·8 nM) S protein was incubated with PCs for 1 h at 37°C, and whole cell protein lysates collected in RIPA buffer as described before.
Assessment of cell viability
Cell viability of cells exposed for 6 and 24 hrs to 1 μg/mL (5·8 nM) S protein was evaluated using the Viability/Cytotoxicity Assay Kit (Biotium #30002), according to manufacturer’s guidelines. Calcein-AM identified live cells, while EthDIII the dead cells. Cells treated with 0.1% saponin for 10 min served as positive control for EthDIII staining. Experiments were performed in duplicates.
Assessment of cell proliferation
The Click-iT EdU Cell Proliferation Kit for imaging (C10337 – ThermoFisher Scientific) was used to assess cell proliferation, according to the manufacturer’s instructions. Cells were incubated with EdU for 24 hrs in the presence of S protein (1 μg/mL – 5·8 nM) and then analyzed. Where required, cells were incubated with the anti-CD147 neutralizing antibody (20 μg/mL). Experiments were performed in duplicates.
2D-Matrigel angiogenesis assay
Human CAECs were seeded on the top of Matrigel (Corning® Matrigel® Growth Factor Reduced Basement Membrane Matrix, cat# 356231) either alone (4,000 cells/well) or in coculture with PCs (4,000 CAECs + 1,500 PC/well), using Angiogenesis μ-Slides (IBIDI, UK) and growth factors free medium. The S protein was added to the system at a concentration of 1 μg/mL (5·8 nM). Images were taken after 5 hrs, and the total tube length per imaging field was measured. To assess the interaction between PCs and CAECs, PCs were labelled with the red fluorescent tracker Vybrant™ DiI Cell-Labeling Solution (Invitrogen). For experiments requiring the CD147 blockade, 100,000 PCs in a total volume of 100 μL were pre-incubated with the anti-CD147 antibody (20 μg/mL) for 1 hour at 20°C. Experiments were performed in triplicates.
Scratch (gap closure) migration assay
A scratch was produced in confluent PCs in the centre of each well. Cells were washed with PBS to remove detached cells and incubated with the experimental ECBM2 medium (FBS and growth factors free). Cell proliferation was inhibited with hydroxyurea (2 mM). Where required, cells were pre-incubated with the anti-CD147 antibody (20 μg/mL) for 1 h at 37°C. The S protein was added to the system at a concentration of 1 μg/mL (5·8 nM). The scratch area was measured at baseline and after 16 hrs. The percentage of wound closure was calculated. Experiments were performed in 96-well plates in 4 to 5 replicates.
Detection of ERK1/2 phosphorylation/activation
For detection of ERK1/2 phosphorylation in cardiac PCs, the ERK1/2 ELISA® Kit was employed, according to the manufacturer’s instructions (Abcam ab176660). PCs were pre-incubated with anti-ACE2 (20 μg/mL, as described before5) or anti CD147 (20 μg/mL) antibodies for 1 hat 37 °C and then exposed to the S protein (500 ng/mL – 2·9 nM) for 1 h at 37°C.
Measurement of cytokines
PCs were maintained for 24 hrs in serum- and growth factors-free medium. Next, cells were incubated with the anti-CD147 antibody for 1 h (20 μg/mL), after which the S protein (1 μg/mL – 5·8 nM) was added for 24 hrs. Then, conditioned media were collected for ELISA measurement of monocyte chemoattractant protein-1 (MCP1/CCL2, R&D DY279), tumour necrosis factor (TNFα, R&D DY210), interferon gamma (IFNγ, R&D DY285B), interleukin-6 (IL-6, R&D DY206), interleukin-1 beta (IL-1β, R&D DLB50), and interleukin-18 (IL-18, R&D DY318). Media were centrifuged at 1,000 g, at 4°C, for 10 min, and stored at -80 °C. Cell protein extracts were collected using RIPA buffer and the total amount of protein was quantified for normalization of secreted factors (BCA assay). Secreted factors are expressed as pg per 100 μg of total cellular protein.
Effect of PC secretome on EC apoptosis
CAECs were seeded in 96-well plates. After 24 hrs, cells were incubated for additional 24 hrs with the PC conditioned medium (collected as described above). PC medium was diluted 1:2 with fresh medium deprived of growth factors, with a final serum concentration of 1%. CAECs were also incubated with the S protein (1 μg/mL – 5·8 nM) for 24 hrs. Cell apoptosis was assessed using the CaspaseGlo 3/7 assay (G8090, Promega, UK) according to the manufacturer’s instructions. Data were expressed as fold changes versus the control group.
Statistics
Data were analyzed using Prism version 8.0 and expressed as individual values and as means ± standard error of the mean. Statistical differences were determined using unpaired T-tests or 1-way or 2-way ANOVAs as appropriate. Non-parametric tests were applied, when appropriate. Significance was assumed when P ≤ 0·05.
Results
Cardiac PCs express ACE2 and CD147
Cardiac PCs express lower levels of both ACE2 and CD147 transcripts compared with cardiac fibroblasts or CAECs used as positive controls (Figure 1A&B). BSG (CD147) transcript in PCs was remarkably higher than ACE2 (Figure 1C). This data was confirmed using immunocytochemistry (Figure 1D&E) and western blotting (Figure 1F).
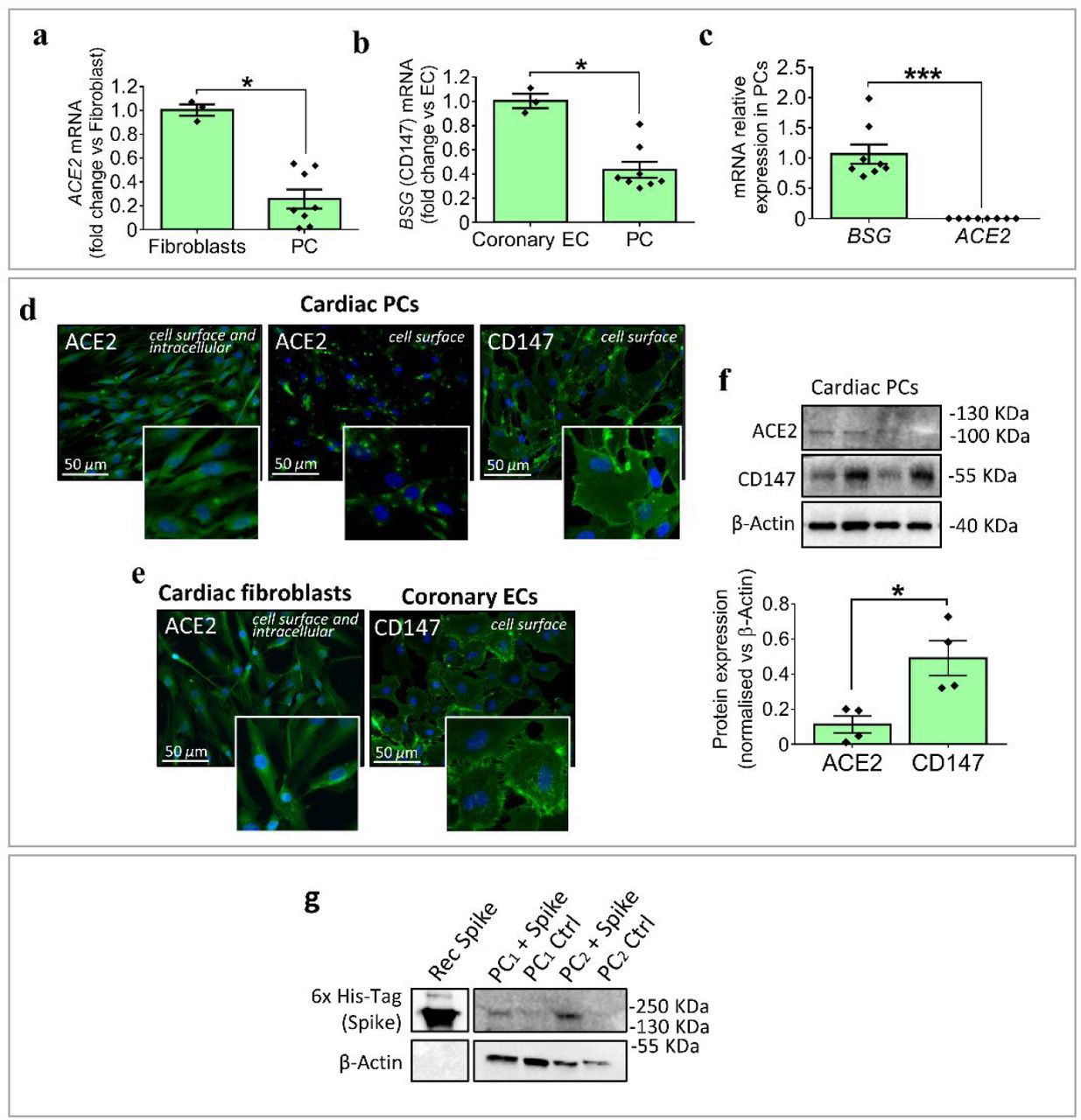
(a&b) Expression of ACE2 and BSG (CD147) mRNA in cardiac PCs, reported as fold change versus cardiac fibroblasts and coronary ECs used respectively as positive controls for ACE2 and BSG. Per each cell population, genes were first normalized against the housekeeping UBC. N=3 EC/fibroblasts, N=8 PC. (c) Relative expression of ACE2 and BSG (CD147) mRNA in cardiac PCs. N=8. (d) Expression of ACE2 and CD147 in expanded cardiac PCs assessed by immunostaining. In green ACE2 and CD147, in blue DAPI (nuclei). (e) Expression of ACE2 and CD147 in cardiac fibroblasts and coronary ECs used as positive controls. (f) Relative expression of ACE2 and CD147 proteins in cardiac PCs, measured using western blotting. N=4. Individual values and means ± SEM. *P < 0·05, ***P < 0·001. (g) Western blotting analysis of PCs (N=2) exposed to the S protein and respective controls. The bands corresponding to the 6x His-Tag recognize the His-tagged S protein. The purified S protein was used as a positive control.
We next verified the binding of S protein, which was tagged with a 6x-HIS sequence for easy detection. Western blotting detected the presence of bands corresponding to the HIS-tagged S protein in PCs exposed to the protein (Figure 1G). The recombinant S protein was used as a positive control.
The SARS-CoV-2 S protein disrupts the pro-angiogenic activity of cardiac PCs
The exposure to S protein for 6 and 24 hrs did not affect PC viability (Figure 2A) and proliferation (Figure 2B). In an in vitro angiogenic assay, the presence of PCs (identified by staining with a red fluorescent dye) increased the formation of CAEC networks (CAEC+PC vs CAEC alone, P<0·0001), with this response being diminished in the presence of the S protein (CAEC+PC spike vs CAEC+PC ctrl, P<0·01); whereas the S protein did not inhibit network formation by CAECs in the absence of PCs (Figure 2D). Moreover, when considering the age of the PC donor, we could appreciate that adult patients-derived PCs were more susceptible to the anti-angiogenic effect of the S protein than paediatric patients-derived PCs (P<0·01) (Figure 2E). Finally, in a scratch assay, the S protein increased the motility of cardiac PCs (spike vs ctrl, P<0·01) (Figure 2F).

(a) Cell viability. Live cell imaging of PCs after 6 and 24 hrs of exposure to the S protein. In green, Calcein-AM shows the cytoplasm of live cells. The red fluorescence of EthD-III indicates the nuclei of dead cells (not detected). Saponin treatment, used as a positive control, shows the nuclear staining of EthD-III in the absence of Calcein-AM. (b) Cell proliferation. Cells were exposed for 24 hrs to the S protein or vehicle in the presence of EdU. Proliferation was measured as the % of EdU+ cells. N=8 PCs. (c-e) Matrigel assay with coronary artery ECs (CAECs) and cocultures of CAEC + PCs. Cells were incubated on the top of Matrigel for 5 hrs, in the presence of the S protein or vehicle. (c) Representative images at the end of the protocol. PCs were labelled with the red fluorescent tracker dil to assess the interaction with ECs (inserts in the bottom row). (d) Graphs report the total tube length per imaging field. (e) Comparison between cocultures of ECs and paediatric and adult patients-derived PCs. N=3 paediatric PCs, N=3 adult PCs. (f) Migration gap closure assay. A scratch was created in confluent PCs and images taken at baseline. Cells were incubated with the S protein or vehicle for 16 hrs and final images were recorded. The area of gap closure was calculated as % of the baseline area. N=5 PCs. For all assays, the SARS-Cov-2 S protein was used at a concentration of 1 μg/mL. Individual values and means ± SEM. *P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001.
S protein-induced effects on cardiac PC function are CD147 dependent
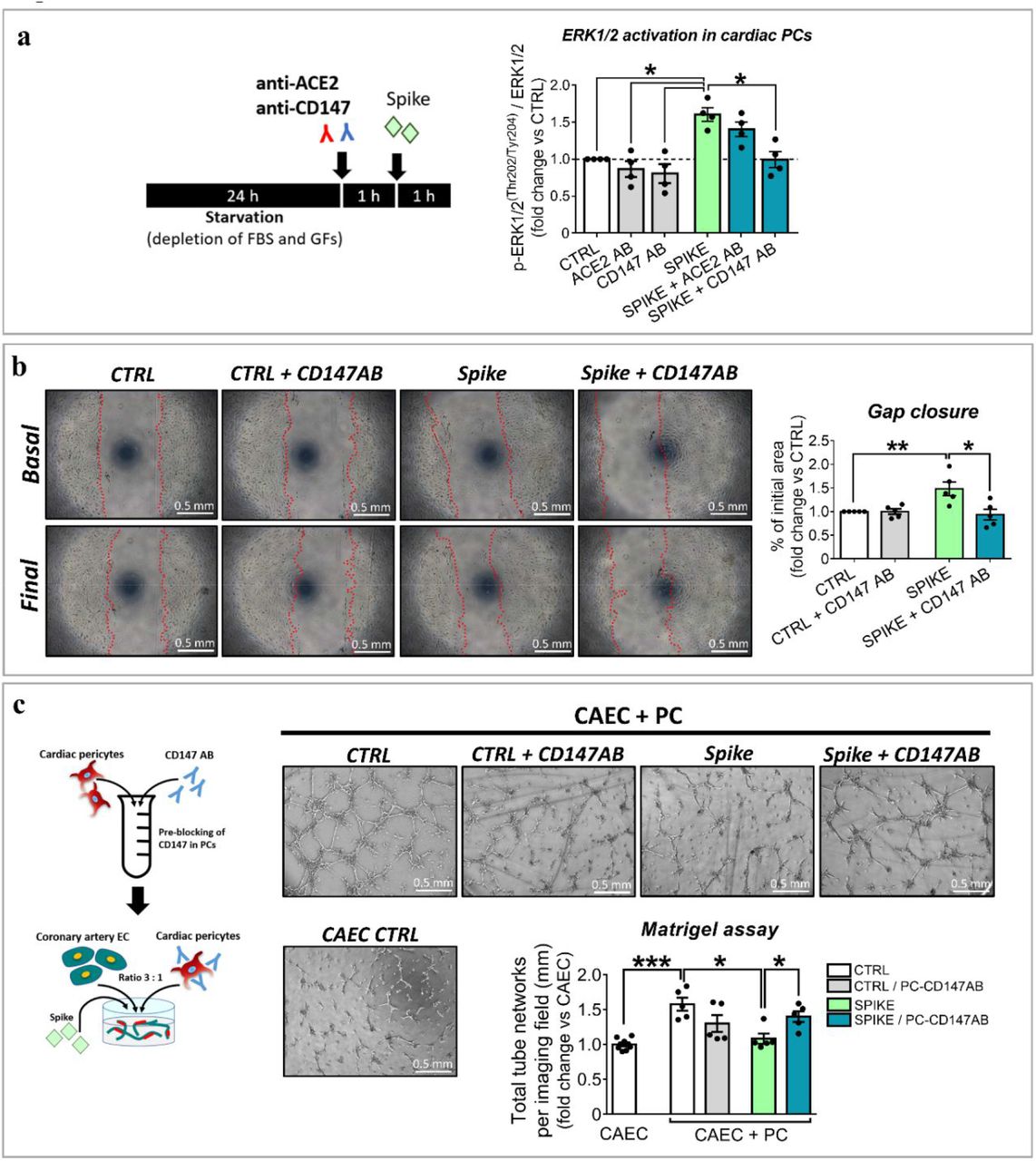
We next investigated the intracellular signalling triggered by the S protein. As shown in Figure 3A, PCs treated with the S protein had significantly increased levels of phospho-ERK1/2 (ratio P-ERK1/2 to total ERK1/2, spike vs ctrl, P<0·05). This response was abolished by a CD147 neutralizing antibody (spike vs spike+CD147AB, P<0·05), but not by an antibody neutralizing ACE2. As shown in Figure 3B, the CD147 blockade also prevented the S protein from inducing PC migration (spike vs spike+CD147AB, P<0·05) and inhibiting PC-CAEC network formation on Matrigel (spike vs spike+CD147AB, P<0·05, Figure 3C).

(a) Intracellular ERK1/2 phosphorylation/activation and role of ACE2 and CD147. Cardiac PCs were kept for 24 hrs without serum and growth factors and then exposed for 1 h to the S protein (500 ng/mL) or vehicle. For receptor blockade, PCs were pre-incubated with antibodies anti-ACE2 or anti-CD147 for 1 h (see cartoon). The bar-graph reports the ratio between phospho-ERK1/2 and total ERK1/2, as measured by ELISA. N=4 PCs. (b) Migration assay. A scratch was created in confluent PCs and images taken at baseline. Where the blockade of CD147 was required, cells were pre-incubated with an antibody anti-CD147 for 1 h. Then, the S protein (1 μg/mL) or vehicle were added to the system for 16 hrs. Final images were recorded. The area of gap closure was calculated as % of the baseline area. N=5 PCs. (c) Matrigel assay with coronary artery ECs (CAECs) and cocultures of CAECs + PCs. Where the blockade of CD147 was required, cardiac PCs were pre-incubated with an antibody anti-CD147 for 1 h. Then, cells were incubated on the top of Matrigel for 5 hrs, in the presence of the S protein (1 μg/mL) or vehicle (see cartoon). The bar-graph indicates the total tube length per imaging field. N=5 PCs. Individual values and means ± SEM. *P< 0·05, **P<0·01, ***P<0·001.
S protein-primed PCs secrete pro-inflammatory cytokines
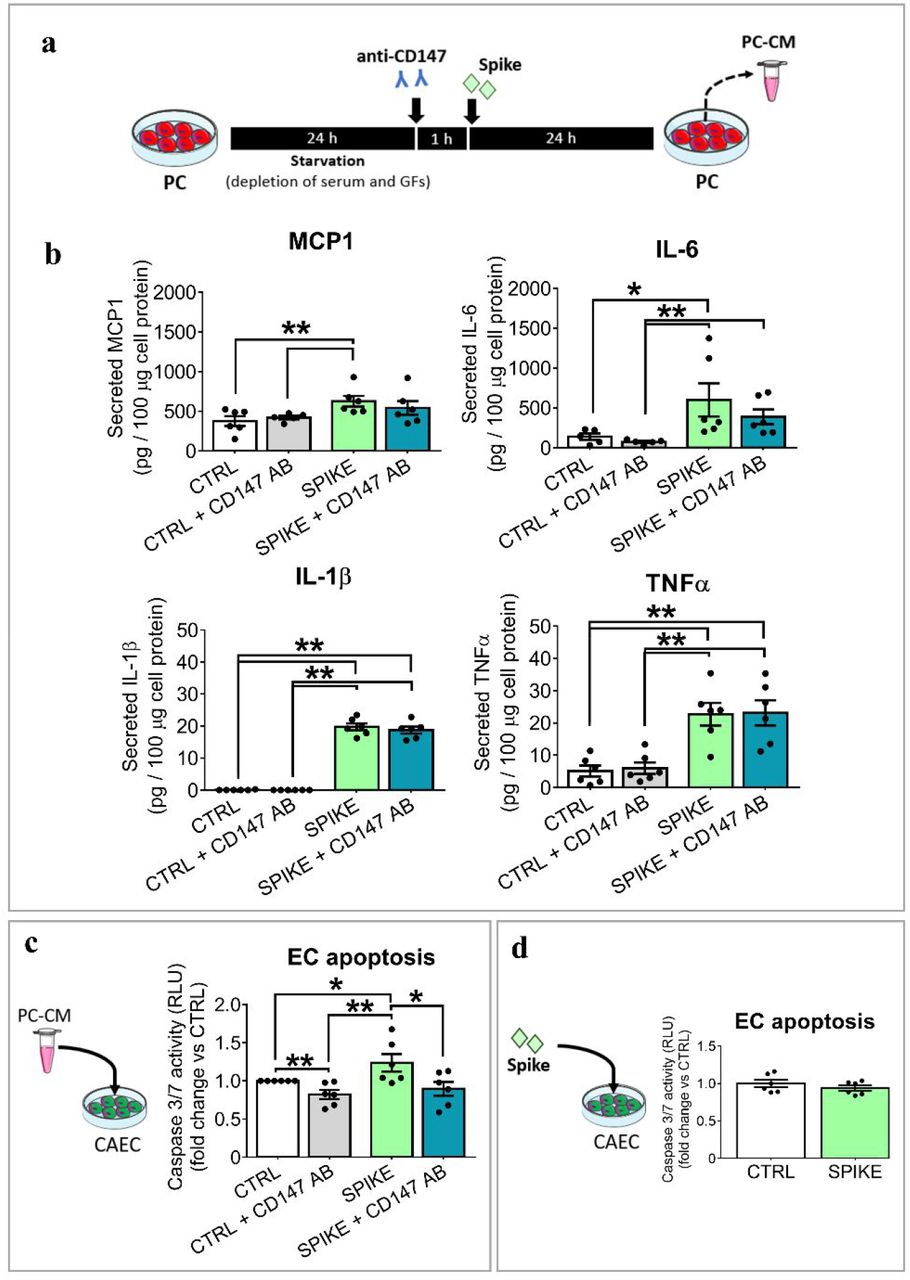
Next, we assessed if the S protein triggers the production of pro-inflammatory cytokines in cardiac PCs. We selected a panel of molecules implicated in the “cytokine storm”.23 As illustrated in Figure 4A&B, the S protein induced PCs to secrete larger amounts of MCP1, IL-6, IL-1β and TNFα (spike vs ctrl, P<0·05), independently of the donor age. The CD147 blocking failed in preventing this alteration (spike vs spike+CD147AB, N.S.). The remaining two cytokines, the pro-inflammatory IL-18 and the anti-inflammatory IFN-γ, were not detectable.

(a) Experimental design to collect the PC conditioned medium (PC-CM). S protein, 1 μg/mL. (b) Secreted factors in the PC-CM normalized against the total cellular proteins. N=5-6 per group. (c) Apoptosis of ECs exposed to the P-CM for 24 hrs. (d) Apoptosis of ECs exposed to S-protein for 24 hrs. In (c&d) values are relative luminescence units (RLU) and are expressed as fold change vs the control group. N=6/group. Individual values and means ± SEM. * P < 0·05, ** P < 0·01.
As shown in Figure 4C, exposure to the media from S protein-primed PCs induced the Caspase 3/7 activity in CAECs (spike vs ctrl, P < 0·05), with this pro-apoptotic effect being reduced by the anti-CD147AB. Conversely, the S protein did not cause direct pro-apoptotic effect on CAECs (Figure 4D).
Discussion
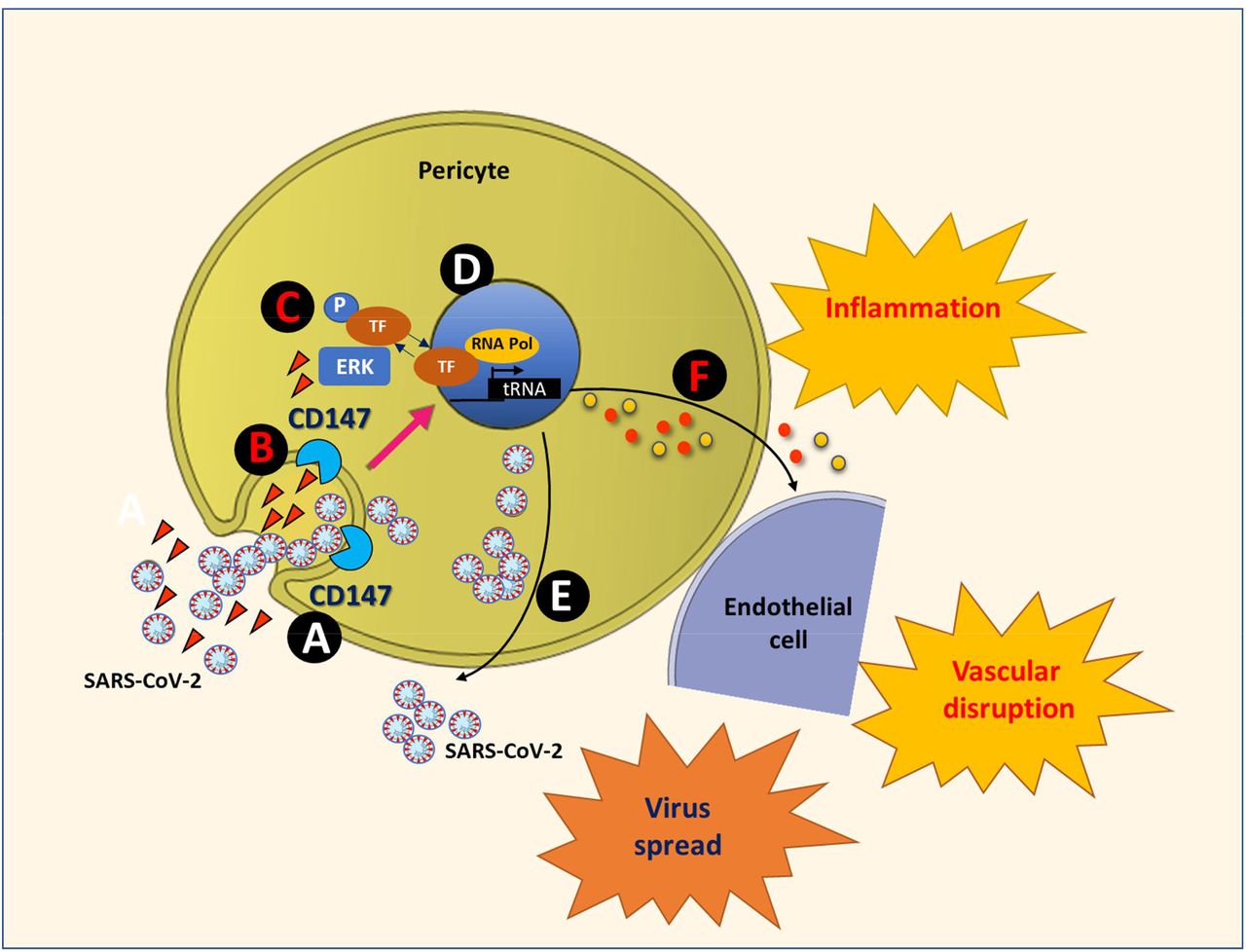
Our study provides novel proof-of-concept evidence for S protein to cause molecular and functional changes in human vascular cells through the CD147 receptor (summarised in Figure 5).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The SARS-CoV-2 Spike proteins, either presented by the virus or as circulating molecules, engage with the CD147 receptor on the cell surface (A). This contact could lead to the internalization of the viral particles/proteins together with the receptor (B), or rather to the activation of CD147 intracellular signalling, with the phosphorylation/activation of ERK1/2 (C). The translocation of phospho-ERK1/2 into the nucleus activates the transcription of target genes (D). Also, phospho-ERK1/2 favours the replication and translation of the viral RNA genome. This leads to the release of new viral particles, supporting the virus spread (E). The transcriptional activation of pro-inflammatory cytokines leads to the production of a pro-inflammatory secretome, which propagates the damage to neighbouring cells (F). The result will be vascular disruption and the onset of microvascular disease.
The SARS-CoV-2 virus particle consists of the three structural proteins: S, membrane (M) and envelope (E), embedded in a lipid bilayer surrounding a helical nucleocapsid comprised of the viral genomic RNA bound to the nucleocapsid (N) phosphoprotein. While the M and E proteins are involved in viral assembly, the S protein mediates cell entry following priming/activation by host cell proteases.5 Moreover, the S protein activates the Raf/MEK/ERK signal transduction pathway in host cells.4 Manipulation of the host ERK1/2 signalling pathway is reportedly instrumental to viral replication,4,24 and to the induction of cyclooxygenase-2, a prostaglandin synthetase involved in inflammation.25 Pharmacological inhibition or knockdown of ERK1/2 by small interfering RNAs suppressed coronavirus replication.24 Therefore, drugs that block the binding of SARS-CoV-2 S protein to cell receptors and/or inhibit downstream signalling pathway may be potential candidates for the treatment of COVID-19.
Our study provides novel insights into the mechanism used by the virus to infect and cause vascular damage. By binding cell receptors, the multifunctional S protein opens the path to the virus entry and manipulates the host intracellular machinery to cause vascular cell dysfunction. The concentration of S protein employed for the in vitro studies is similar to those of other ligands known to activate ERK1/2 in PCs (EGF – 0.83 nM, and bFGF – 0.6 nM, former data from our group).18 Activation of PC migration and inhibition of interactive cooperation between PCs and CAECs in angiogenesis assays are both suggestive of a dysfunctional phenotype. In fact, the detachment of PCs from the perivascular compartment results in vulnerable capillaries that are prone to instability, pathological angiogenesis, and, ultimately, rarefaction. On the other hand, the S protein did not impinge upon PC viability. SARS-CoV-2 cannot replicate without the machinery of a host cell. It could be therefore counterproductive for the virus to kill the cell at the initial stage of the S protein engagement with host receptors. The S protein also activated or enhanced the production of pro-inflammatory cytokines by cardiac PCs. MCP1, IL-6, IL-1β and TNFα are typical components of the “cytokine storm” associated with high mortality in COVID-19 patients.23 This pro-inflammatory secretome could produce harmful paracrine effects on the surrounding vascular cells, as our experiment on CAEC apoptosis suggests. This mechanism can propagate functional alterations even to those cell populations which may not be directly infected by the virus, ultimately contributing to vascular disruption.
A recent report showed that the full S1 subunit causes the phosphorylation/activation of MEK in human pulmonary vascular cells.7 However, using only the ACE2 RBD failed to do so, therefore it was not clear if the signalling started from the ACE2 receptor.7 The authors suggested that an alternative receptor different from ACE2 might mediate the signalling of the S protein in vascular cells. Basigin/CD147, a plasma membrane protein associated with oligomannosidic glycans, has emerged as a novel receptor for SARS-CoV-2.9 Initial evidence from studies in lung epithelial cells and immune cells26 has been contradicted by an investigation on CD147-transfected HEK 293 cells.27 These discrepancies may be reconciled with the requirement of coreceptors for the effective binding of the S protein to CD147. Importantly, an open-label, clinical trial of meplazumab, a humanized therapeutic monoclonal antibody against CD147, showed striking improvements in COVID-19 patients.28 Our study confirms the data from single-cell sequencing studies showing cardiac PCs express the ACE2 mRNA transcript.11,12 However, ACE2 protein expression was less than CD147. Moreover, only CD147 blockade restrained the S protein from inducing ERK1/2 phosphorylation and rescued several functional features of PCs that were compromised by the S protein, including PC stability and pro-angiogenic activity. Interestingly, the anti-angiogenic effect of the S protein was more pronounced in PCs from adult patients compared with PCs from paediatric patients. Epigenetic mechanisms associated with the naivety of the youngest heart could account for this difference. Finally, CD147 blockade protected CAECs from the paracrine apoptotic action of S protein-primed PCs but failed to prevent the induction of multiple pro-inflammatory cytokines, thus suggesting the latter phenomenon involves mechanisms unrelated to the CD147 receptor.
Most persons infected with SARS-CoV-2 display an antibody response to the S protein between day 10 and day 21 after infection. This initial stage is crucial for SARS-CoV-2 to cause damages, some of which could be attributed to the S protein acting as a pleiotropic ligand that induces the cell molecular machinery in favour to the virus. The three most advanced vaccines (from Oxford/AstraZeneca, Pfizer/BioNTech and Moderna) all work by inducing human cells to make copies of the S protein. The expression of the vaccinal S protein is likely limited to the site of muscular injection. However, the accidental passage of the adenoviral vector or mRNA-containing nanoparticles into the circulation may lead to systemic expression, which could be harmful in predisposed individuals. Reported side effects, including fatigue, muscle and joint pain, headache, severe fever, and anaphylactic reactions are likely to be due to a transient reaction to the vehicle. However, more investigation is necessary to determine if the occurrence of side effects may be associated with systemic levels of the transduced S protein.
Source of funding
This work was funded by the Elizabeth Blackwell Institute (EBI) Rapid Response COVID-19 award “COVID-19 S-protein binding to ACE2 negatively impacts on human cardiac pericyte function – a mechanism potentially involved in cardiac and systemic microvascular failure” to PM. In addition, it was supported by the British Heart Foundation (BHF) Centre for Regenerative Medicine Award (II) – “Centre for Vascular Regeneration” (RM/17/3/33381) to PM (co-lead of WP3).